
Timisoara_Med 2021, 2021(2), 2; doi:10.35995/tmj20210202
Case Report
Dual Cancer in a Patient with Type 1 Gaucher Disease: Case Report and Literature Review
1
Department of Gastroenterology and Hepatology, “Victor Babes” University of Medicine and Pharmacy, 300041 Timisoara, Romania; popa.alexandru@umft.ro (A.P.); roxanasirli@gmail.com (R.S.)
2
Department of Anatomy and Embryology, “Victor Babes” University of Medicine and Pharmacy, 300041 Timisoara, Romania; nicoiacob@yahoo.co.uk
*
Correspondence: patricia.lupulescu@yahoo.com; Tel.: +40-742892603
How to cite: Popa, A.; Lupulescu, P.; Iacob, N.; Sirli, R. Dual Cancer in a Patient with Type 1 Gaucher Disease: Case Report and Literature Review. Timisoara Med. 2021, 2021(2), 2; doi:10.35995/tmj20210202.
Received: 18 May 2021 / Accepted: 10 October 2021 / Published: 26 October 2021
Abstract
:Gaucher Disease (GD) is a rare inherited lysosomal storage disease caused by biallelic mutations in the gene encoding the lysosomal enzyme acid β-glucocerebrosidase, with progressive accumulation of the sphingolipid glucosylceramide in the lysosomes of macrophages, mainly in the liver, spleen, bone, and bone marrow. It is characterized by anemia, thrombocytopenia, hepatosplenomegaly, sometimes severe bone involvement and, in certain forms, neurological involvement. We present the case of a 77-year-old male, diagnosed in 2006, at 63, with GD type1. At the onset, the patient presented fatigue, asthenia, upper abdominal discomfort, skin pallor and minor bone pain. During follow-up, CT was performed and displayed an enlarged spleen (25 cm) and a well-defined mass in the kidney cortex, but it could not establish whether it was malignant or not. Considering the patient refused surgery at that moment, a new CT exam was performed six months later, and the kidney tumor significantly increased in size. Nephrectomy and necessity splenectomy were performed, and recovery was uneventful. The histopathological examination revealed clear cell renal cell carcinoma, grade 2 Fuhrman. Enzyme substitution therapy with Imiglucerasum was initiated in 2012 with favorable evolution. Seven years later, during a routine ultrasound follow-up, a 4/3.8 cm hyperechoic mass was found in the eighth liver segment. The biological tests revealed a high value of alpha-fetoprotein (AFP-185.2 ng/mL). Considering the patient’s history, possible diagnoses of the newly found focal liver lesion were: gaucheroma, metastasis secondary to kidney cancer, or a primitive liver tumor. Core biopsy was performed, and it was suggestive of a poorly differentiated hepatocellular carcinoma G3. A non-anatomical resection of liver segments VII and VIII with partial resection of inferior vena cava was performed, followed by full post-operative recovery. A histopathological examination confirmed the initial diagnosis. Currently, one month postoperatively, the patient in good clinical condition, continues the enzyme replacement treatment and is undergoing oncological follow-up. We presented a complex case of GD, diagnosed at an advanced age, who responded well to substitution therapy in regard to hematologic and bone symptoms, but who unfortunately developed two types of cancer managed aggressively despite the advanced age.
Keywords:
Gaucher Disease; hepatocarcinoma; renal carcinoma; malignancyIntroduction
Gaucher Disease (GD) is the most common lysosomal storage disease, with a global prevalence estimated at 1:50,000–1:100,000 [1]. In Romania, GD is believed to be extremely rare. Seventy-nine patients have been reported with a prevalence of 1/250,000. More than 300 GBA mutations have been described in the GBA1 gene [2]. Very rarely, GD can also be caused by a deficiency in the glucocerebrosidase activator, saposin C [3].
GD is characterized by anemia, thrombocytopenia, hepatosplenomegaly, sometimes severe bone involvement and, in certain forms, neurological involvement. The irregularity in GD’s clinical presentations may be explained by the extensive range of phenotypes [4]. The initial symptoms vary considerably. Almost half of the patients experience fatigue. Growth retardation and delayed puberty are frequent in early life. Splenomegaly is observed in more than 90% of patients and is sometimes massive, causing abdominal discomfort with resultant pancytopenia caused by hypersplenism [5]. Bone involvement causes acute pain manifested as very painful “bone crises”, frequently limited to one joint and can be accompanied by fever and leukocytosis with an aseptic blood culture [6]. The infiltration of the lungs by Gaucher cells seems to be related to pulmonary involvement in GD patients, creating an interstitial disease that can lead to pulmonary fibrosis [7,8]. Rarely, renal involvement can also appear, glomerular infiltration by Gaucher cells can cause proteinuria and hematuria [9].
The diagnosis of GD is frequently made several years after the first clinical and laboratory signs. It requires an enzymatic assay to evaluate glucocerebrosidase activity. Bone marrow aspiration is not mandatory; however, it can be useful in patients with isolated thrombocytopenia or splenomegaly. Acid β-glucocerebrosidase gene (GBA1) mutations located on chromosome 1q21 are diagnostic of GD. More than 400 mutations have been described in the GBA1 gene [10,11,12].
Enzyme replacement therapy (ERT) which helps substitute the defective or deficient beta-glucocerebrosidase and substrate-reduction therapy (SRT) which reduces glycolipid accumulation by decreasing the synthesis of glucocerebroside (the substrate of the deficient enzyme) represent Gaucher-specific therapy. Splenectomy, hematopoietic stem cell transplantation, gene therapy and enzyme-enhancement therapy represent other treatment options [13].
This paper aims to present a unique case of an elderly patient diagnosed with type 1 GD who presented two independent cancers during follow-up.
Case Report
Patient Information and Initial Assessment
We present the case of a 77-year-old male, diagnosed in 2006, at 63, with GD type 1. At the onset (2006), the patient presented progressive fatigue, asthenia, upper abdominal discomfort, skin and mucosal pallor as well as minor bone pain, mainly of the lumbar spine. The symptoms started 6 months before diagnosis. The physical examination discovered massive hepatosplenomegaly and multiple ecchymoses. The laboratory tests revealed the presence of anemia (Hb: 9.7 g/dL), thrombocytopenia (45.000/mm3), and elevated serum ferritin.
Diagnosis of GD
The patient was referred to the hematology department, where, after excluding other common causes of anemia, a bone marrow biopsy was performed. It revealed the presence of cells with an eccentric nucleus and abundant cytoplasm with a striated appearance, resembling wrinkled tissue paper, suggestive of Gaucher cells (Figure 1).
The definitive diagnosis was based on the enzymatic level (B-glucocerebrosidase—0.94 nmol/h/mg < 15% of normal; Chitotriozidase—9750 nmol/h/mL plasma 1.8xN) and on genetic testing (genotype N370S/R463C). At that moment, specific treatment with GD was not started due to economic constraints.
Diagnosis and Management: Renal Carcinoma
In 2007, during follow-up, abdominal ultrasound revealed an enlarged spleen, about 25 cm, with several hypoechoic nodules. A 3.5 cm hypoechoic well-defined mass was visualized in the cortex of the left kidney. Cholelithiasis was also present. Computer tomography was performed and displayed a well-defined mass in the kidney cortex, but it could not establish whether it was malignant or not. It also showed massive splenomegaly (25/21/18 cm) with multiple round, solid, hypodense, hypovascularized splenic nodules up to 3 cm in diameter (Figure 2). Considering that the patient refused surgery at the moment and the possibility that the kidney mass is nothing more than an accumulation of Gaucher cells or a hemangioma, it was decided to delay surgical treatment of the kidney tumor.
A new CT exam was performed six months later, and compared with the previous examination, the kidney tumor significantly increased in size (with approximately 8 mm) (Figure 3).
Nephrectomy and necessity splenectomy was performed and recovery after surgery was uneventful. The histopathological examination revealed clear renal cell carcinoma, grade 2 Fuhrman (Figure 4). The splenic parenchyma included numerous bulky macrophages with discrete eosinophilic cytoplasm, fibrillated vacuolar appearance with small, dark, eccentric, small nuclei (Gaucher cells) (Figure 5).
During follow-up, in the absence of specific enzyme replacement therapy, fatigue, anemia and bone pain worsened. The patient presented increasing bone pain. The bone MRI revealed medullary infiltration and osteolytic lesions, Düsseldorf score 5, class 2b. (Figure 6). Bone densitometry was used to diagnose osteopenia.
ERT Therapy Initiation and Follow-Up
Enzyme replacement therapy with Imiglucerasum (30 UI/kg) was initiated in 2012 and the patient’s evolution was favorable, although splenectomy and nephrectomy were performed. Six years after the therapy initiation, the patient’s overall condition improved significantly, the hemoglobin and platelets normalized, bone damage improved, and bone pain disappeared. However, osteodensitometry (DEXA) showed a decrease in bone density, and a mild form of pulmonary hypertension appeared (Table 1). Due to associated cardiac disease, Eliglustat could not be administrated.
Diagnosis and Management: Hepatocellular Carcinoma
Seven years after the initiation of therapy, although the patient overall condition improved significantly, in September 2019 during a routine ultrasound follow-up, a 4/3.8 cm hyperechoic mass was found in the eighth liver segment (Figure 7). The biological tests revealed a high level of ferritin (823 ug/L) and a high value of alpha-fetoprotein (AFP-185.2 ng/mL). Additionally, LS values by TE revealed the presence of advanced liver fibrosis (8.9 kPa).
Considering the patient’s history, possible diagnoses of the newly found focal liver lesion were Gaucheroma and metastasis secondary to kidney cancer or a primitive liver tumor.
Contrast enhanced ultrasound (CEUS) was performed. The lesion showed hyperenhancement in the arterial phase (Figure 8), being completely filled at second 20. Wash-out was observed beginning in the portal phase, which becomes marked in the late phase (Figure 9). The conclusion of the CEUS examination was a malignant tumor of the liver.
MRI was also performed. It showed a well-defined mass located in segment VIII of the right hepatic lobe, hyperenhanced in the arterial phase, with diffusion restriction, in close contact with the inferior vena cava and with the right hepatic vein. Slight wash-out was observed in the parenchymal phase, while in the hepatospecific phase, the lesion was hypoenhanced as compared to the surrounding liver. The enhancement pattern was suggestive of a hypervascularized malignant tumor (Figure 10).
CEUS and MRI were inconclusive to differentiate between a primitive liver tumor or a metastasis. Thus, core biopsy was performed (Figure 11), which revealed the presence of large, atypical cells, with pale eosinophilic cytoplasm, round and oval hyperchromic nuclei, with moderate pleomorphism and mitotic activity suggestive of a poorly differentiated hepatocellular carcinoma G3.
Considering the patient’s good clinical condition and the HCC localization, a non-anatomical resection of liver segments VII and VIII with partial resection of inferior vena cava was performed, followed by full post-operative recovery. The histopathological examination of the resected surgical specimens confirmed once again the diagnosis of poorly differentiated hepatocellular carcinoma G3. One month postoperatively, the patient in good clinical condition, continues the enzyme replacement treatment and is undergoing oncological follow-up.
Discussion
Clinically, GD has been classified into three types based on the presence or absence of early-onset neurologic involvement: type 1 (non-neuronopathic), type 2 (acute neuronopathic) and type 3 (chronic neuronopathic), in 92%, 1% and, respectively, 7% of patients. The majority of patients (74%) are diagnosed with GD before age 30 (mean age, 19 y) [14]. This case’s particularity is that our patient was diagnosed at an advanced age, in contrast to the mean age of diagnosis.
While GD is predominately an illness that manifests in infancy, the symptoms can appear at any time from infancy to old age. Due to a wide variability in clinical manifestations and severity of non-specific symptoms, diagnosis is challenging. This is exacerbated by the low experience among physicians with the early disease features because of its low incidence [15]. Numerous patients may experience significant delays in obtaining a definitive diagnosis.
A study published by Mistry et al. showed that there is a delay between the onset of symptoms and diagnosis for up to 10 years in GD, and that the patients consulted a mean number of 8 physicians (internists, hematologist, pediatricians, gastroenterologist, etc.) before a diagnosis was agreed upon [16]. Another study revealed that numerous patients faced diagnostic delays and misdiagnoses, 1 in 6 patients affirming that they were not diagnosed with GD for 7 years or more after first referring a physician. Cytopenia, splenomegaly and bone pain were the most common symptoms at first presentation in both studies [15]. Given all of these data, diagnosis can be even more problematic when dealing with an elderly patient, as in our case.
In the past, splenectomy was performed for therapeutic reasons in cases of severe hypersplenism. Current practice is that splenectomy must be avoided since it is linked to a more severe disease progression and many complications. It is a risk factor for decrease life expectancy, bone complications, cirrhosis, and pulmonary complications. Additionally, some studies revealed that splenectomy enhances the risk of malignancy. In our case splenectomy could not be avoided, ten years after the splenectomy the patient developed a slight form of pulmonary hypertension and osteoporosis, but normalization of platelet count was obtained after introduction of ERT [17].
Lately, many studies have shown that GD is correlated with an increased risk of malignancies. Unfortunately, the determining factors of malignancy are not completely known in a subset of patients with GD1. Several factors could contribute to cancer development in GD, including polarization of macrophages to the alternatively activated phenotype, chronic inflammation, chronic B-cell stimulation, splenectomy, hyperferritinemia, lysosomal dysfunction, and endoplasmic reticulum stress. Recent studies have highlighted T-cell dysfunction and modifier genes contributing to an increased cancer risk in GD. From all cancer types, hematological malignancies seem to be associated more frequently with GD. The risk of multiple myeloma is estimated at 37-fold in comparison to the general population. Some studies have also suggested increased cancer risk for non-hematological malignancies, such as hepatocellular carcinoma, renal cell carcinoma [17,18]. Our patient had no hematological malignancy. however, he developed renal cell carcinoma, followed ten years after by hepatocellular carcinoma.
Almost 50 % of GD1 patients have focal lesions which are located in the spleen or in the liver. They are most likely “gaucheromas”, but differential diagnosis has to be made also with malignant lesions such as hepatocellular carcinoma, lymphoma of the spleen or other neoplasia. The imaging characteristics of gaucheromas are not typical. Consequently, it is challenging to differentiate it from other lesions [19,20].
Hepatocellular carcinoma (HCC) represents about 90% of primary liver cancers [21]. It is associated in 90% of the cases with a known underlying condition, most frequently cirrhosis of various etiologies, such as chronic viral hepatitis, chronic alcohol abuse, metabolic diseases such as NAFLD, genetic haemochromatosis or in some cases, alpha-1-antitrypsin deficiency [22]. Overall, one-third of cirrhotic patients will develop HCC during their lifetime [23].
The extent of liver damage in GD still represents a topic of debate. GD patients have an increased risk for hemosiderosis, focal liver fibrosis, steatosis, overt cirrhosis. [24,25]. In a study made on a group of 37 GD type 1 adults, the prevalence and predictors of liver fibrosis was evaluated by vibration-controlled transient elastography (TE). It was found that 19 % of the patients had significant fibrosis. Significant fibrosis was associated with splenectomy and with scores and biomarkers of GD severity [26]. Another study that included 31 patients showed that there is a strong correlation between liver stiffness (LS) values obtained from Magnetic Resonance Elastography and GD-DS3 score (severity score) [27]. It is currently unknown which patients are at risk for developing HCC, but studies have shown that this risk can be increased by the presence of fibrosis and cirrhosis. [27]. In our patient, LS values by TE revealed the presence of advanced liver fibrosis (8.9 kPa).
Because of the higher incidence and prevalence of cancer, a prophylactic approach is required, even in a patient with a good course of GD. Gaucher patients should consult their clinical reference center for specific measures of prophylaxis as regular determination of biomarker levels (chitotriosidase, CCL18 and ferritin), AFP, PSA, high resolution abdominal ultrasound, immune electrophoresis, liver elastography, routine colonoscopy after 50 years of age. Some centers perform whole-body MRI, which represents the most appropriate method to assess the morphology and dimensions of the liver and spleen. [28].
In our case, although the patient was carefully monitored and received proper therapy for GD, he developed two distinct neoplasms ten years apart, a renal cell carcinoma and a poorly differentiated hepatocellular carcinoma.
Conclusions
We presented a complex case of Gaucher disease, diagnosed at an advanced age, who responded well to substitution therapy in regard to hematologic and bone symptoms, but who unfortunately developed two types of cancer managed aggressively despite the advanced age. We suggest a prophylactic approach is required in all GD patients.
Author Contributions
Conceptualization, A.P. and R.S.; Methodology, A.P.; Software, P.L.; Validation, N.I., A.P. and R.S.; Formal Analysis, R.S.; Investigation, A.P., N.I., R.S.; Resources, A.P.; Data Curation, P.L.; Writing—Original Draft Preparation, A.P.; Writing—Review and Editing, A.P., P.L., R.S.; Visualization, A.P., P.L., R.S.; Supervision, R.S.; Project Administration, A.P.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chen, M.; Wang, J. Gaucher disease: Review of the literature. Arch. Pathol. Lab. Med. 2008, 132, 851–853. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E. Gaucher disease: Complexity in a “simple” disorder. Mol. Genet. Metab. 2004, 83, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Kaloterakis, A.; Filiotou, A.; Koskinas, J.; Raptis, I.; Zouboulis, C.; Michelakakis, H.; Hadziyannis, S. Systemic AL amyloidosis in Gaucher disease. A case report and review of the literature. J. Intern. Med. 1999, 246, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Bucerzan, S.; AlKhzouz, C.; Nascu, I.; Zimmerman, A.; Popp, R.; Lazea, C.; Grigorescu-Sido, P. OC-87 Gaucher disease in romania—Baseline characteristics, specific diagnosis. treatment and outcome. Arch. Dis. Child. 2017, 102, A34–A35. [Google Scholar] [CrossRef]
- Stone, D.L.; Ginns, E.I.; Krasnewich, D.; Sidransky, E. Life-threatening splenic hemorrhage in two patients with Gaucher disease. Am. J. Hematol. 2000, 64, 140–142. [Google Scholar] [CrossRef]
- Pastores, G.M.; Meere, P.A. Musculoskeletal complications associated with lysosomal storage disorders: Gaucher disease and Hurler-Scheie syndrome (mucopolysaccharidosis type I). Curr. Opin. Rheumatol. 2005, 17, 70–78. [Google Scholar] [CrossRef]
- Amir, G.; Ron, N. Pulmonary pathology in Gaucher’s disease. Hum. Pathol. 1999, 30, 666–670. [Google Scholar] [CrossRef]
- Mistry, P.K.; Sirrs, S.; Chan, A.; Pritzker, M.R.; Duffy, T.P.; Grace, M.E.; Meeker, D.P.; Goldman, M.E. Pulmonary hypertension in type 1 Gaucher’s disease: Genetic and epigenetic determinants of phenotype and response to therapy. Mol. Genet. Metab. 2002, 77, 91–98. [Google Scholar] [CrossRef]
- Santoro, D.; Rosenbloom, B.E.; Cohen, A.H. Gaucher disease with nephrotic syndrome: Response to enzyme replacement therapy. Am. J. Kidney Dis. 2002, 40, E4. [Google Scholar] [CrossRef]
- Koprivica, V.; Stone, D.L.; Park, J.K.; Callahan, M.; Frisch, A.; Cohen, I.J.; Tayebi, N.; Sidransky, E. Analysis and classification of 304 mutant alleles in patients with type 1 anti type 3 gaucher disease. Am. J. Hum. Genet. 2000, 66, 1777–1786. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Gahl, W.A. Lysosomal storage diseases. Metab. Dis. Found. Clin. Manag. Genet. Pathol. 2017, 2, 1–70. [Google Scholar] [CrossRef] [PubMed]
- Stirnemann, J.Ô.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A review of gaucher disease pathophysiology, clinical presentation and treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef]
- Revel-Vilk, S.; Szer, J.; Mehta, A.; Zimran, A. How we manage Gaucher Disease in the era of choices. Br. J. Haematol. 2018, 182, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, N.J.; Goldblatt, J.; Villalobos, J.; Charrow, J.; Cole, J.A.; Kerstenetzky, M.; Vom Dahl, S.; Hollak, C. Long-term clinical outcomes in type 1 Gaucher disease following 10 years of imiglucerase treatment. J. Inherit. Metab. Dis. 2013, 36, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Belmatoug, N.; Bembi, B.; Deegan, P.; Elstein, D.; Göker-Alpan, Ö.; Lukina, E.; Mengel, E.; Nakamura, K.; Pastores, G.M.; et al. Exploring the patient journey to diagnosis of Gaucher disease from the perspective of 212 patients with Gaucher disease and 16 Gaucher expert physicians. Mol. Genet. Metab. 2017, 122, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Sadan, S.; Yang, R.; Yee, J.; Yang, M. Consequences of diagnostic delays in type 1 Gaucher disease: The need for greater awareness among Hematologists-Oncologists and an opportunity for early diagnosis and intervention. Am. J. Hematol. 2007, 82, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Taddei, T.; vom Dahl, S.; Rosenbloom, B.E. Gaucher disease and malignancy: A model for cancer pathogenesis in an inborn error of metabolism. Crit. Rev. Oncog. 2013, 18, 235–246. [Google Scholar] [CrossRef]
- Shiran, A.; Brenner, B.; Laor, A.; Tatarsky, I. Increased risk of cancer in patients with gaucher disease. Cancer 1993, 72, 219–224. [Google Scholar] [CrossRef]
- Tseng, S.-Y.; Niu, D.-M.; Chu, T.-H.; Yeh, Y.-C.; Huang, M.-H.; Yang, T.-F.; Liao, H.-C.; Chiang, C.-C.; Ho, H.-C.; Soong, W.-J.; et al. Very rare condition of multiple Gaucheroma: A case report and review of the literature. Mol. Genet. Metab. Rep. 2019, 20, 100489. [Google Scholar] [CrossRef] [PubMed]
- Regenboog, M.; Bohte, A.E.; Somers, I.; van Delden, O.M.; Maas, M.; Hollak, C.E.M. Imaging characteristics of focal splenic and hepatic lesions in type 1 Gaucher disease. Blood Cells Mol. Dis. 2016, 60, 49–57. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 183–263. [CrossRef]
- Fitzmaurice, C.; Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzman, N.; Amoako, Y.; et al. The burden of primary liver cancer and underlying etiologies from 1990 to 2015 at the global, regional, and national level results from the global burden of disease study 2015. JAMA Oncol. 2017, 3, 1683–1691. [Google Scholar] [CrossRef]
- Chen, S.L.; Morgan, T.R. The natural history of hepatitis C virus (HCV) infection. Int. J. Med. Sci. 2006, 3, 47–52. [Google Scholar] [CrossRef]
- Adar, T.; Ilan, Y.; Elstein, D.; Zimran, A. Liver involvement in Gaucher disease—Review and clinical approach. Blood Cells Mol. Dis. 2018, 68, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, F.; Dalla Salda, A.; Carubbi, F. Energy balance, glucose and lipid metabolism, cardiovascular risk and liver disease burden in adult patients with type 1 Gaucher disease. Blood Cells Mol. Dis. 2018, 68, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, F.; Cassinerio, E.; Dalla Salda, A.; Motta, I.; Bursi, S.; Donatiello, S.; Spina, V.; Cappellini, M.D.; Carubbi, F. Prevalence and predictors of liver fibrosis evaluated by vibration controlled transient elastography in type 1 Gaucher disease. Mol. Genet. Metab. 2018, 125, 64–72. [Google Scholar] [CrossRef]
- De Fost, M.; Vom Dahl, S.; Weverling, G.J.; Brill, N.; Brett, S.; Häussinger, D.; Hollak, C.E.M. Increased incidence of cancer in adult Gaucher disease in Western Europe. Blood Cells Mol. Dis. 2006, 36, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, N.J.; Aggio, M.C.; Andersson, H.C.; Andria, G.; Charrow, J.; Clarke, J.T.R.; Erikson, A.; Giraldo, P.; Goldblatt, J.; Hollak, C.; et al. Gaucher disease type 1: Revised recommendations on evaluations and monitoring for adult patients. Semin. Hematol. 2004, 41, 15–22. [Google Scholar] [CrossRef]
Figure 1.
Bone marrow biopsy—cells with an eccentric nucleus and abundant cytoplasm with a striated appearance, resembling wrinkled tissue paper, suggestive of Gaucher cells.
Figure 1.
Bone marrow biopsy—cells with an eccentric nucleus and abundant cytoplasm with a striated appearance, resembling wrinkled tissue paper, suggestive of Gaucher cells.

Figure 2.
Computer tomography showing massive splenomegaly (25/21/18 cm) with multiple round, solid, hypodense, hypovascularized splenic nodules up to 3 cm in diameter.
Figure 2.
Computer tomography showing massive splenomegaly (25/21/18 cm) with multiple round, solid, hypodense, hypovascularized splenic nodules up to 3 cm in diameter.
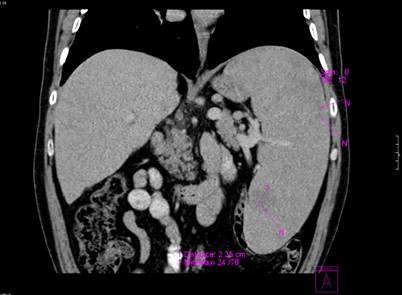
Figure 3.
Computer tomography showing massive splenomegaly and a well-defined mass in the kidney cortex significantly increased in size compared with the previous examination.
Figure 3.
Computer tomography showing massive splenomegaly and a well-defined mass in the kidney cortex significantly increased in size compared with the previous examination.

Figure 4.
Histopathological examination (hematoxylin and eosin staining) of the kidney tumor revealing clear renal cell carcinoma grade 2 Fuhrman.
Figure 4.
Histopathological examination (hematoxylin and eosin staining) of the kidney tumor revealing clear renal cell carcinoma grade 2 Fuhrman.

Figure 5.
Histopathological examination (hematoxylin and eosin staining) of the spleen revealing numerous bulky macrophages with discrete eosinophilic cytoplasm, fibrillated vacuolar appearance with small, dark, eccentric, small nuclei (Gaucher cells).
Figure 5.
Histopathological examination (hematoxylin and eosin staining) of the spleen revealing numerous bulky macrophages with discrete eosinophilic cytoplasm, fibrillated vacuolar appearance with small, dark, eccentric, small nuclei (Gaucher cells).

Figure 6.
Bone MRI revealing medullary infiltration and osteolytic lesions, Düsseldorf score 5, class 2b.
Figure 6.
Bone MRI revealing medullary infiltration and osteolytic lesions, Düsseldorf score 5, class 2b.
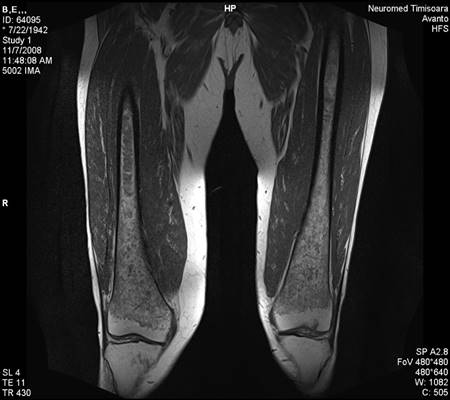
Figure 7.
Abdominal ultrasound showing 4/3.8 cm hyperechoic mass in the eighth liver segment.

Figure 8.
CEUS image showing hyperenhancement of the tumor in the arterial phase.

Figure 9.
CEUS image showing wash-out that started in the portal phase.

Figure 10.
MRI showing a well-defined mass located in segment VIII of the right hepatic lobe hyperenhanced in the arterial phase, with diffusion restriction, in close contact with the inferior vena cava and with the right hepatic vein.
Figure 10.
MRI showing a well-defined mass located in segment VIII of the right hepatic lobe hyperenhanced in the arterial phase, with diffusion restriction, in close contact with the inferior vena cava and with the right hepatic vein.

Figure 11.
Ultrasound image captioned during core—biopsy.


Table 1.
Main clinical and paraclinical characteristics after substitution therapy initiation.
| Year | 2012 | 2016 | 2017 | 2018 | 2019 |
|---|---|---|---|---|---|
| Clinic | Asthenia Bone pain | Good overall condition | Good overall condition | Good overall condition | Good overall condition |
| RBC | 10 g/dL | N | N | N | N |
| Ferritin | 1370 | 1090 | 917 | 823 | |
| MRI | Bone marrow infiltration Dusseldorf score 5 Class Terek 2b | Dusseldorf score 2 Class Terek 1a | Dusseldorf score 2 Class Terek 1a | Dusseldorf score 2 Class Terek 1a | No data |
| DEXA | Lumbar spine—1.010 Left femur—0.845 | Lumbar spine—1.021 Left femur—0.848 | Lumbar spine—1.069 Left femur—0.858 | Lumbar spine—0.9 Left femur—0.849 | Lumbar spine—0.86 Left femur—0.79 |
© 2021 Copyright by the authors Licensed as an open access article using a CC BY 4.0 license